

")





Blogs: 15 Месяцев: миодистрофия Дюшенна — рожденный бегать бежать не может ☹

15 месяцев: миодистрофия Дюшенна
— рожденный бегать бежать не может ☹
Информационно-просветительский гуманитарный проект «15 месяцев» — это цикл материалов о необычных людях — пациентах с редкими (орфанными) болезнями, о которых не написано в студенческих учебниках. Считается, что вероятность встретить на профессиональном пути редкого пациента у обычного врача ничтожно мала, поэтому в академических аудиториях им не уделяют должного внимания, что в повседневной жизни приводит к диагностическим ошибкам, упущенному времени и поломанными судьбами и жизням.
Научные редакторы проекта:
- Роман Деев (СЗГМУ им. И.М. Мечникова, Артген - https://artgen.ru/) ;
- Алексей Паевский (http://neuronovosti.ru/, https://med-history.livejournal.com/).
Миодистрофия Дюшенна — тяжёлая наследственная мышечная дистрофия; родители могут ее заметить достаточно рано — с первых шагов ребенка или, когда их малышу исполнится в среднем, от 2 до 5 лет. Мышечная слабость быстро прогрессирует, лишает ребенка, а затем и взрослого человека привычной нам физической активности, без лечения эта болезнь в 100% случаев заканчивается смертью. А как сложится судьба пациента если изобрести самое современное и самое совершенное лекарство — генную терапию? Мы пока этого не знаем, но уже сейчас первые препараты для генной терапии начинают использовать для лечения таких детей.
При этой болезни первыми поражаются — а значит, погибают, мышцы тазового пояса и бедер, затем мышцы верхнего плечевого пояса, а также сердце и дыхательная мускулатура. Мы знаем, что при этой болезни нередко поражается головной мозг, костная ткань и гладкая мускулатура ЖКТ [1]. Следовательно, пациенты оказываются в инвалидном кресле в тот период, когда яркая жизнь только начинается — в возрасте около 10-12 лет, и с каждым новым днем мальчики чувствуют нарастающую слабость, зная, что уже никогда не будут похожи на своих сверстников.
Частота развития этого заболевания составляет от 3,5 до 5,5 на 5 000 новорожденных мальчиков, может поражать целые семьи, что позволяет считать миодистрофию Дюшенна одной из самых частых болезней среди редких наследственных заболеваний [2].
История изучения
Начало изучения заболевания приходится на первые десятилетия XIX века. В то время был крайне распространен туберкулез, который проявлялся как в различных типичных, так и нетипичных формах. Первое упоминание о характерной патологии скелетных мышц датируется 1830 годом, когда английский хирург, анатом и физиолог Чарльз Белл описал случай 18-летнего пациента с выраженной мышечной атрофией и быстро прогрессирующей физической слабостью, которая впервые проявилась в возрасте 10 лет [3].
 Чарлз Белл, английский хирург, анатом и физиолог.
Чарлз Белл, английский хирург, анатом и физиолог.
© Wikimedia Commons.
Затем в 1834 году итальянский врач Джованни Семмола представил случай заболевания двух мальчиков, у которых с первых лет жизни наблюдалась увеличение размеров отдельных групп мышц, принятое врачом за основное проявление болезни [4].
В 1836 году заболевание продолжили описывать соотечественники Семмолы — Гаэтано Конте и Л. Джойя, которые представили случай двух братьев со стремительно развивающейся слабостью в совокупности с уменьшением мышечной массы бедер и увеличением икроножных мышц. Болезнь манифестировала в возрасте 8–10 лет; один из пациентов умер от нарушения работы сердца [5].
Следующее упоминание о заболевании приходится на 1853 год и связано с именем английского хирурга Уильяма Джона Литтла, работы которого внесли новые данные в изучение миодистрофии: им при вскрытии было обнаружено замещение большей части мышечных волокон жировой тканью [6]. Это позволило с точностью описывать патологию термином «миодистрофия».
До 1852 года учеными велись споры о природе миодистрофии — противостояли мнения о поражении самих волокон мышцы и о нарушении их иннервации. Наконец, в 1852 году английским врачом Эдвардом Мерионом была проведена аутопсия, в результате которой было установлено отсутствие явных признаков поражения нервных проводников. Э. Мерион предложил гипотезу о мышечной первопричине болезни [7]. После публикации нескольких работ Мериона эта миодистрофия некоторое время носила его имя, однако в настоящий период за ней закрепилось имя французского невролога-электрофизиолога Гийома Дюшенна.
В 1861 и 1868 годах Гийом Дюшенн представил клинические наблюдения пациентов с этим заболеванием, в том числе с данными электрической стимуляции мышечных и нервных волокон. По полученным электрофизиологическим данным он охарактеризовал патологию не только как «миодистрофию», но и как «гипертрофический паралич» [8].

Гийом Дюшенн, французский невролог-электрофизиолог.
© Wikimedia Commons.
Причина развития заболевания
Причиной заболевания являются мутации в гене белка дистрофина (DMD), расположенном в коротком плече Х-хромосомы (локус Xp21). Этот ген, судя по всему — самый большой ген человека с очень запутанной молекулярной организацией. В гене как минимум пять промоторов и 79 экзонов, он кодирует громадный белок с молекулярной массой в 427 килодальтон.
Место расположения гена предполагает бессимптомное носительство мутантного гена женщинами, а при его передаче — проявление в фенотипе лишь у сыновей. Дело в том, что в женском организме мутировавший ген на одной из Х-хромосом компенсируется второй, «здоровой», Х-хромосомой. Проявление мутации у девочек встречается очень редко — 1:50 000 000 новорожденных девочек (для развития должна иметься аномалия гена в обеих Х-хромосомах) [9]. При наследовании мутантной Х-хромосомы мальчиками, в организме которых генетически материал представлен одной Х и одной Y-хромосомой, не происходит компенсация нарушения, и заболевание развивается в 100% случаев.
В основе всех нарушений при миодистрофии Дюшенна лежит дефицит белка дистрофина, функция которого заключается в поддержании нормальной, достаточной связи структур внутри мышечных волокон — цитоскелета, со структурами внеклеточного матрикса.
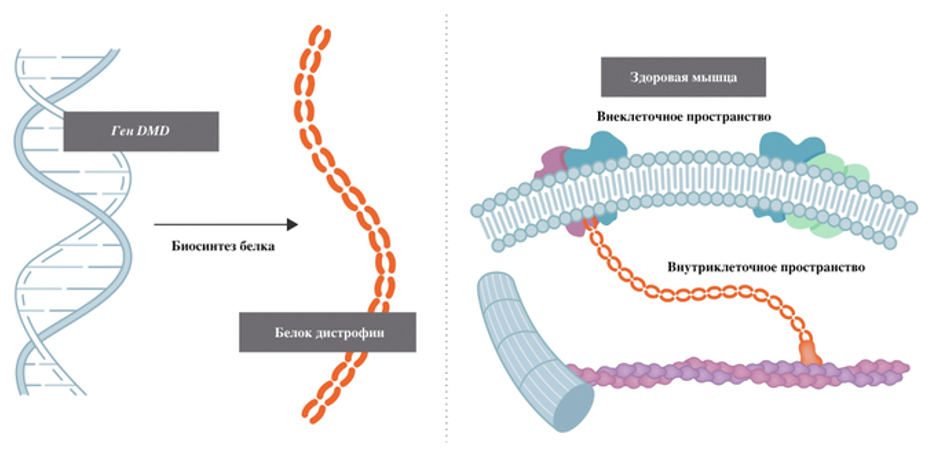
Белок дистрофин и его функция в норме (переработанное изображение).
При отсутствии дистрофина проницаемость мембран волокон возрастает, что сопровождается выходом из клеток креатин-фосфокиназы (КФК) (специфичного маркера мышечного повреждения) и развитием воспаления. Эти процессы приводят к гибели клеток: поперечнополосатая мышечная ткань замещается жировой и соединительной. Кроме того, важную роль играют иммунопатологические механизмы, сопровождающие хроническое воспаление: измененные мембраны мышечных волокон воспринимаются иммунной системой организма как чужеродные [10].
Клинические проявления
Первые признаки болезни могут быть определены уже в возрасте 2–5 лет, когда быстрая утомляемость ребенка, нежелание играть, постоянное желание быть на руках у взрослых часто не настораживают родителей. Беспокойство родителей появляется в тот момент, когда становится заметным изменение походки («утиная» — широко расставленные ноги и передвижение на цыпочках, «генеральская» — переразгибание в поясничной области), затрудненный подъем по лестнице, неумение прыгать, увеличение и уплотнение икроножных мышц [1].

Ранние клинические проявления миодистрофии Дюшенна
(DOI: 10.5075/epfl-thesis-9104 с изм.).

Миодистрофия Дюшенна характеризуется быстро развивающейся деформацией скелета в поясничном, грудном отделах, изменениями в костях стоп, утратой подвижности суставов. Потеря способности к самостоятельному передвижению возникает в возрасте 10–12 лет.
К концу второго — началу третьего десятков лет пациенты оказываются зависимы от посторонней помощи для выполнения самых простых ежедневных действий. Слабость крупных мышц сопровождается утратой сухожильных рефлексов (например, коленного, ахиллового), поражением мимической мускулатуры, низкорослостью, умственной отсталостью, разрушением костей из-за отсутствия физической активности и приема глюкокортикостероидов.

Наряду с этим на поздних стадиях пациенты перестают не только самостоятельно ходить, но и дышать — в связи с поражением дыхательных мышц пациентам необходима респираторная поддержка сначала в дневное, а затем и в ночное время. При обычном течении заболевания смерть наступает чаще всего из-за дыхательной и сердечной недостаточности в возрасте около 26 лет [11]. Повреждение сердца происходит на фоне гипо- и адинамии пациента, поэтому часто остается нераспознанным и нелеченым, пока не достигает стадии необратимых изменений [12]. Кроме этого, в сердце развиваются специфические воспалительные процессы, разрастание соединительной ткани, гипертрофия. В итоге это приводит к перерастяжению камер сердца — сердце становится похоже на растянутый мешочек, который не может обеспечить весь организм достаточным кровообращением. Кроме того, авторы отмечают наличие жизнеугрожающих нарушений ритма [13].
В то же время существует фенотип миодистрофии Дюшенна, характеризующийся доброкачественным течением, описанный П.Э. Беккером. При миодистрофии Беккера дистрофин синтезируется в малых количествах, либо происходит синтез аномального белка, способного к частичному выполнению своих функций [14]. Миодистрофия Беккера медленно прогрессирует — пациенты сохраняют способность к самостоятельной ходьбе в течение 15–20 лет от начала заболевания.
Даже не все врачи знают, что миодистрофия Дюшенна часто поражает и когнитивную сферу. Примерно каждый пятый — каждый третий пациент страдает когнитивными расстройствами разной тяжести. Почему же так происходит? До сих пор существуют разные предположения.
Так, например, работа исследователей из Санкт-Петербурга обращает внимание на то, что белок дистрофин входит в состав дистрофин-гликопротеинового комплекса (ДГК), который присутствует не только в мышечных клетках, но и нейронах, участвуя в работе потенциалзависимых каналов и формировании синапсов. Кроме того, судя по всему, некоторые мутации в гене дистрофина влияют и на уровень одного из нейротрофинов — фактора роста головного мозга (ФРГМ), что в итоге приводит к недостаточному росту и дифференцировке нейронов.
Диагностика заболевания
В настоящее время диагностика миодистрофии Дюшенна основана на тщательном неврологическом осмотре, оценке уровней особого фермента — креатинфосфокиназы (КФК), применении электрофизиологического метода — электронейромиографии (ЭНМГ), биопсии, а также молекулярно-генетического исследования; причем последнее имеет решающее значение в достоверной верификации проблемы со здоровьем у мальчика.
В 100% случаев для мужского пола и в 50% для женского уровень КФК при фенотипе Дюшенна повышается в 10 раз и более, при фенотипе Беккера — в 5.
ЭНМГ позволяет различить первичное поражением мышц при миодистрофии Дюшенна и вторичное — при поражения нервной системы, связанное с другими заболеваниями.
При биопсии скелетных мышц определяются погибшие и гипертрофированные волокна, кроме того, визуализируются жировые отложения и разрастание соединительной ткани. Возможно применение иммуногистохимического метода оценки биоптатов мышц с использованием моноклональных антител к дистрофину.
Для количественного определения дистрофина используется вестерн-блоттинг с денситометрией — сочетание реакции дистрофина и моноклональных антител с разделением белков по фазам в специальном геле с точным определением количества дистрофина (возможно обнаружение до 1 нанограмма). Генетический анализ включает в себя поиск аномалий гена DMD как до — у матери, так и после рождения — у ребенка [15].
Лечение заболевания
Исцеляющего лечения, к сожалению, нет. Человечество его еще не придумало.
Медикаментозная терапия включает в себя классическую схему лечения и генную терапию. Из применяемых лекарственных средств эффективными оказываются глюкокортикостероиды (ГКС), однако только на начальных этапах прогрессирования мышечной атрофии — они способны замедить воспалительное повреждение мышц; при утраченной способности к передвижению ГКС бесполезны. Исследования показывают, что ГКС продлевают длительность ходьбы, уменьшают выраженность сколиоза и улучшают сердечно-легочную функцию. При этом следует понимать: эта группа препаратов не устраняет причину заболевания — генетический дефект и связанный с ним дефицит дистрофина, а лишь снижает сопутствующие некрозу мышц иммунопатологические процессы и уменьшает выраженность кислородной недостаточности мышечной ткани. В связи с этим терапия ГКС увеличивает мышечную силу лишь в краткосрочной перспективе (от 6 месяцев до 2 лет). Доза применяемых препаратов не имеет стандартизации: существуют разные схемы приема, однако все они связаны с развитием побочных эффектов различной степени тяжести, прямо пропорциональной длительности применения. Справляться с ними приходится путем введения дополнительных лекарственных средств, направленных на коррекцию ожирения, задержки физического развития, язв желудка и кишечника, расстройств переваривания-всасывания питательных веществ, разрушения костей из-за остеопороза, подавления иммунной защиты организма [16].
Перспективным методом лечения миодистрофии Дюшенна является применение геннотерапевтических методов, цель которых — устранение причины заболевания, то есть мутации в гене DMD. 14 ноября 2019 года в Ньюкасле (Великобритания) состоялась встреча более 100 представителей ведущих центров изучения нервно-мышечных болезней, групп защиты интересов пациентов, национальных и местных аптек, а также финансируемых государством организаций, работающих над внедрением генной терапии в рутинную практику лечения миодистрофии Дюшенна.
ААВ — аденоасоциированные вирусы находятся в центре внимания разработчиков генной терапии миодистрофии Дюшенна неслучайно. Они вызывают слабый иммунный ответ и, в отличие от некоторых других вирусных векторов (например, аденовирусных), не интегрируют свой генетический материал в геном хозяина, оставаясь в виде небольших эписомальных частиц в ядре, отдельно от ДНК пациента. Это делает их более безопасными с точки зрения потенциальных побочных мутагенных эффектов. Однако рААВ векторы имеют ограниченную емкость и могут нести генетический материал размером до 4,7 тысяч пар нуклеотидов (т.п.н.). Транскрипт DMD имеет кодирующую последовательность длиной 14 т.п.н., значительно превышая емкость рААВ [17].
Проблема большого размера гена DMD отчасти решается переносом наиболее важных его доменов, обеспечивающих максимальную функцию транслируемого белка (т.н. мини- и микродистрофина) при наименьшем размере. Тем не менее, важно признать, что даже наиболее функциональные укороченные версии дистрофина не так эффективны, как полноразмерный белок, и лишь трансформируют злокачественное течение миодистрофии Дюшенна в более мягкий фенотип — миодистрофию Беккера. Неясной остается длительность выработки микродистрофина у пациентов с миодистрофией Дюшенна, получавших генную терапию ААВ векторами. Точную временную границу провести не представляется возможным из-за непрерывно текущих процессов дегенерации и регенерации мышечных волокон [18].
У исследователей возникает другой вопрос. В какой момент введение геннотерапевтической конструкции будет оптимальным? Если ждать, пока пациент станет старше, чтобы задействовать в продукцию микродистрофина единовременно как можно большее количество мышечных волокон, процесс дегенерации будет уже запущен. В свою очередь, если вводить генный препарат очень маленьким детям, из-за увеличения количества и объема мышечных волокон по мере взросления будет происходить «разбавление» способных к продукции микродистрофина волокон. Повторное введение вирусного вектора при этом потенциально опасно: насколько бы ни был слабым иммунный ответ, вызываемый рААВ, он неминуемо будет запущен после первичного введения [17].
Массовое внедрение генной терапии миодистрофии Дюшенна в рутинную практику невозможно без учета и производственных реалий: для эффективной вирусной трансфекции необходимо около 100 триллионов векторных геномов на килограмм массы тела пациента, что требует определенных технологических мощностей. Возможным решением проблемы производства, иммуногенности и инсерционного мутагенеза является применение невирусных векторов: перенос ДНК при помощи физических методов (создание временных дефектов мембран клеток) или катионных липидов/полимеров. В целом, однако, эффективность трансфекции при использовании невирусных методов остается на относительно низком уровне [19].
Может показаться, что генная терапия на данный момент всё ещё является чем-то далёким от обычной практики, витающим на страницах научных журналов. Это не совсем так. В 2016 году FDA (Food and Drug Administration, CША) одобрило Этеплирсен — первый генный препарат для лечения миодистрофии Дюшенна, несущий в себе аминокислотную последовательность, которая связывается с мутировавшим экзоном 51 (наиболее частое место возникновения делеций и дупликаций) и исключает его из синтеза дистрофина. После этого дистрофин начинает синтезироваться в усеченном функциональном виде. Также существуют препараты, предназначенные для пропуска экзона 53 (Голодирсен, Вилтоларсен). Препарат 2а талурен нарушает распознавание точечных мутаций, преждевременно останавливающих синтез дистрофина (10-15% случаев заболевания), позволяя восстановить синтез дистрофина [20]. Первыми критериями эффективности применяемых препаратов являются улучшающиеся, по сравнению с началом лечения, результаты таких тестов, как тест на время вставания (Time to Stand Test), тест на время бега/ходьбы на 10 метров (TTRW), мышечная сила, измеренная ручным динамометром и другие подобные. Кроме клинических данных, оцениваются результаты применения всех методов диагностики в динамике. Для каждого препарата срок достижения желаемых результатов разный, однако результаты оцениваются, начиная с первых дней применения для исключения нежелательных побочных реакций.
Еще одним препаратом для лечения миодистрофии Дюшенна является Эзутромид — модулятор продукции утрофина, аналога дистрофина, выполняющего его функции в эмбриональном периоде. Сейчас препарат находится на 2 фазе клинических испытаний и по их окончании может стать новой ступенью лечения миодистрофии Дюшенна [21].

Текст: Мария Савельева, Роман Деев, Алексей Паевский
КАК ПОЛЮБИТЬ?
Семья Свешниковых уникальна: в ней определенно один за всех, и все за одного. Алексей страдает миодистрофией Дюшенна, он хорошо учится и обожает художественную литературу. У Даши синдром Дауна, она любит играть в теннис и отлично танцует. Родители решились усыновить этих прекрасных детей, чтобы подарить им любовь и создать комфортные условия жизни. Они — муж и жена - основали благотворительный фонд помощи пациентам «МойМио», подают пример своими победами и неудачами и просто радуются каждому дню.
ВСЕГО ЧЕТЫРЕ ГОДА
Мы с мужем много занимались благотворительностью. Я долгое время была волонтером в фонде «Подари жизнь»[1], а муж ездил и помогал детским домам и домам престарелых. Однажды в интернете он увидел пост, в котором рассказывали про мальчика Лешу, который страдает редким заболеванием, и врачи на тот момент пророчили ему всего четыре года жизни. Мы решили, что психологически и финансово готовы подарить ребенку четыре года в нормальной семье, в любви и заботе. Ему было пять, когда мы впервые с ним познакомились. Через четыре года мы смеялись и говорили: «Все, договор исчерпан, время прошло, возвращаем»! Вот уже десять лет, как он у нас в семье.
Когда мы его забирали, он совсем не разговаривал. Только издавал звуки: «би-би», «ням-ням» и жестикулировал. А к семи годам уже потихонечку начал читать. Сейчас в свои 15 лет он говорит все еще немного странно, по-своему: иногда не может полностью сформулировать мысль. Мы до сих пор с ним решаем математические задачки за третий класс. Конечно мы не осилим математику всей десятилетки; мы пытаемся дать ему что-то элементарное, чтобы он мог разбираться во времени, что-то самостоятельно купить и правильно посчитать сдачу в магазине. Пока мы все еще путаемся, сколько минут в часе, сколько месяцев в году. Все это он знает, но все у него в голове находится как будто в каше — в беспорядке, из которого надо постоянно вытягивать информацию. Поэтому у нас каждый день — это уроки-уроки-уроки!

Он очень любит историю и литературу, очень много читает, причем достаточно серьезные книги. Перечитал всего Жуль Верна, Гюго, сейчас второй раз перечитывает «Всадника без головы» Майн Рида. С другими хобби у нас, конечно, довольно туго. Я всегда пытаюсь поддержать любой его интерес, но он у него быстро гаснет: компьютер и «Майнкрафт» - это наше все!

А ВДРУГ Я НЕ СМОГУ ЕГО ПОЛЮБИТЬ?
Вспоминаю, что когда увидела Лешу в первый раз, то очень испугалась. Он был такой страшненький, детдомовский лопоухий мальчик в шортиках и колготках, растянутых на коленках. У него было плохое настроение, он не умел говорить и просто на всех рычал.
Я вышла оттуда и, садясь в машину, подумала: «А вдруг я не смогу его полюбить?». Потом он лег в больницу, где я провела с ним все время госпитализации. Этот период очень сильно нас сблизил. Когда мы поехали домой и в первый раз ехали в лифте, у меня были мысли: «Неужели он не боится? Он едет с незнакомыми дяденькой и тетенькой неизвестно куда»!? Потом поняла, что у него было отношение к нам и нашему дому, как к временному пристанищу. Он никогда никому не был нужен; своей семьи, и своего угла у него не было. Это отношение сохранялось несколько лет. Первое время он всех вокруг называл «мамой». Это травма детского дома. У него вообще очень тяжелая история. Сначала биологическая мама Леши его везде забывала. Потом он попал в детский дом, откуда его взяли в семью. Через полгода выяснилось, что он болен и его сдали обратно. У него постоянно была смена лиц и помещений. Он привык к такому отношению, что его могут вернуть в любой момент. Только позже мы постепенно стали ощущать, как между нами возникает семейная химия.
Десять лет назад я, конечно, была более восторжена. Я очень хотела ребенка и брала пример со своих друзей, у которых тоже дети с различными ограничениями. Тогда Леша еще хорошо ходил, мы много путешествовали. Сейчас все меняется, он уже падает и не может встать сам. Начали проявляться проблемы со здоровьем. Понимаю, что дальше они будут только увеличиваться; назад не повернуть. Внутренне я все прекрасно понимаю. Мы пытаемся решать проблемы по мере их поступления. Мы просто живем, просыпаемся, улыбаемся и делаем какие-то дела. Живем в тех условиях, в которых мы сейчас можем жить.
В свои пятнадцать лет Леша очень сохранный для миодистрофии Дюшенна. В его возрасте с таким заболеванием дети уже сидят в кресле, а он еще ходит. В школу он, правда, ездит на электрической коляске. За ним по расписанию приезжает специальная машина, которая возит Алексея в школу и обратно.
С ГЛАЗ ДОЛОЙ И ИЗ СЕРДЦА ВОН
В России со школой для Алеши было как-то тяжело.
Сначала мы пошли в замечательную школу для инвалидов в Москве. Там есть доступная среда[2], маленькие классы, специальные машины, которые возят детей. Но для нас это было очень далеко, до нее нам нужно было ехать 2,5 часа. К тому же мне приходилось все уроки там сидеть: 40 минут и 20 минут перемена. Все мамы там сидели. Мы проводили в школе вместе с дорогой, по 11 часов и так пять дней в неделю. В какой-то момент мы с Лешей поняли, что это не для нас, поэтому мы перешли на онлайн-обучение. Но в силу своего возраста и, возможно, ментальных особенностей, он не мог просто сидеть и слушать учителя, поэтому мне также же приходилось все время быть рядом.
Во второй класс мы пошли в обычную школу с доступной средой. И тут начались проблемы. Там был лифт, но как им пользоваться никто не знал. Включить его на постоянной основе они не могли, потому что дети будут баловаться и застрянут. Всю первую четверть мы с Лешей поднимались по лестнице, ждали там, чтобы потом его спустить. Затем они наконец нашли «специального человека», который бегал включал и выключал лифт, когда Леше это было нужно.
В классе им никто не занимался. Сначала его посадили на последнюю парту у двери - «с глаз долой и из сердца вон». Он сидел на уроках и ничего, естественно, не понимал, потому что не успевал за всеми. Делать ему было нечего, он то рисовал на пеналах, то на руках. Приходил с уроков весь исписанный. Я ходила в школу разбираться, как на работу. Мы предлагали им разные варианты решения, но школа не шла на контакт. Только через год были сделаны специальные классы, куда Леша ходил на математику и русский. Закончилось наше знакомство с новой школой в один из новогодних праздников.
Ребенок очень сильно хотел пойти на торжество и даже выучил стихотворение, подошел к этому ответственно, ждал этого дня… Потом я узнала, что все дети — его одноклассники, были на празднике, а Лешу учительница в это врем отвела к логопеду (чтобы не мешал). Вот тут я и сломалась.
Забрала документы и решила обучать его на дому.
ФРАНЦИЯ
Каждый год мы посещали различные международные конференции по миодистрофии Дюшенна, общались с людьми и врачами из разных стран. Я узнала про благотворительную ассоциацию «Telethon» во Франции. Они рассказывали, что у них «дети с Дюшенном» живут дольше сорока лет. Это поразило меня: как же так!? Ведь в России, если посмотреть по регионам, ребята умирают в возрасте до двадцати.
Стало ясно, что у меня есть два варианта: оставить Лешу в России и тогда непонятно, что будет дальше, а есть вариант переехать. Я бы осталась воевать за него в нашей стране и дальше, но у этих детей попросту нет столько времени.
Сейчас во Франции он ходит в обычную школу, но в специальный класс. С языком у него, конечно, плоховато, но учителя обучены работать с детьми-иностранцами. Здесь очень заинтересованы в том, чтобы Алеша находился в обществе со всеми и не ощущал себя изгоем. Он рассказывает: «В России я сидел один, в школе никто мной не интересовался. А тут я сижу, ко мне все подходят, со мной все общаются». У него даже девочка была, с которой он сердечками обменивался. Такое же отношение и к Даше — моей дочке (у нее синдром Дауна), все дети бросаются ее обнимать и всегда ведут ее играть вместе. В детском лагере у них постоянно была борьба за то, кто же пойдет с Дашей за ручку. Вероятно, должно пройти еще несколько поколений, чтобы в России массово воспитать в детях такое же отношение друг к другу.
Здесь, во Франции, очень серьезно развита поддержка родителей. Леша как-то раз упал, и этого было достаточно для того, чтобы через пару дней нам привезли специальный подъемник. Медицинскую кровать дали сразу, матрас дают индивидуально и меняют его каждые три года. Коляски тоже подбираются индивидуально, не нужно выискивать каких-то специалистов, физических терапевтов. А когда у нас сломалась коляска, технический специалист приехал тут же, забрал ее и привез отремонтированную через неделю. Все это входит в стандарт оказания помощи, все привозят на дом. Также есть специальные люди, кураторы, которые помогают решать различные документационные и технические вопросы. Семьи здесь имею право на домашнюю помощь. У нас есть соседка, она мать-одиночка ребеночка с особенностями, так вот к ней приходят три раза в неделю и гуляют с ребенком. Каждое утро приходят и помогают ему встать, а вечером помыться и лечь. Помимо всего, родители детей-инвалидов получают аналог зарплаты и трудовой стаж, а не просто пособие и пенсию. Зарплата высчитывается на основании того, в какой степени ребенок способен обслуживать себя самостоятельно и сколько времени родители тратят на уход.
Каждый первый врач тут знает о существовании этого заболевания. Есть кураторы, которые ведут Лешу, а не я самостоятельно звоню и записываю его к врачам. Куратор следит, чтобы Алексей каждые полгода проходил диагностическое обследование. Я получаю письма на почту с информацией о времени записи к врачам, которые проводят все необходимые анализы в течение дня.
ФОНД
Постепенно у нас зарождалась идея создать благотворительный фонд в России. Когда мы забирали Лешу, то, конечно, стали ближе знакомиться с этим заболеванием и узнавать все больше информации. Тогда мы познакомились с Еленой Шепард, соучредителем Фонда и моей подругой. У нее тоже мальчик с Дюшенном. Она давно уже в этом «варилась» и знала ситуацию лучше нас. Все родители жили как-то сами по себе и общались редко. Мы решили: «Надо что-то сделать, чтобы всех объединить».
В четырнадцатом году была организована новогодняя елка. Все прошло настолько хорошо, что мы продолжили развивать эту тему. Следующим шагом было создание летнего лагеря. Первые два сезона мы провели, когда еще не было никакого фонда. Сами находили деньги и искали людей, которые могли бы нам помочь. Туда приехали родители с детьми из разных регионов России и врачи, которые рассказывали об этом заболевании. И вот информация стала передаваться дальше по сарафанному радио, все постепенно расширялось и через год родился фонд «МойМио»[3].

У Фонда несколько программ. Клиника в России, где проводится обследование детей у знающих врачей, летние лагеря и сбор благотворительных средств. Проект «Профессия», который помогает деткам получить образование; юристы, которые защищают права пациентов. Также мы занимаемся просветительской работой: выездные семинары и обучение врачей на местах. Сообщества родителей стали формироваться по регионам. Такое движение в России действительно было нужно, ведь заболевание не такое уж и редкое: семей с миодистрофией Дюшенна около 3-4 тысяч. А большинство врачей и клиник до сих пор не до конца понимают и знают, как грамотно проводить обследование и лечение. До 15-го года, например, о Дюшенне в Москве знали только две больницы.
Меня радует, что в последнее время об этой проблеме больше стали говорить. Должна быть государственная заинтересованность в том, чтобы включить такие заболевания в образовательную программу. Худо-бедно в Москве и в Петербурге можно на метро куда-то приехать и с третьего раза свой диагноз получить. А как быть в Дагестане или Якутии? И тем не менее, в Москве нет ни одного нервно-мышечного центра, в то время как во Франции их 23, но она то маленькая по сравнению с Россией. Сами врачи должны быть заинтересованы обучаться и идти на контакт. Проблема не в том, что они плохие врачи, а в том, что об этом не написано в учебниках. Пациента с таким редким заболеванием не успеешь встретить на недельном цикле в мединституте. Мне кажется, что новое поколение врачей больше хочет развиваться и будет более внимательным к проблемам наших семей.
Подробнее узнать о заболевании и для перевода помощи следует обратиться в Благотворительный фонд «МойМио».
Литература
- Suthar R., Sankhyan N. Duchenne Muscular Dystrophy: A Practice Update. Indian J Pediatr. 2018;85:276–281. doi: 10.1007/s12098-017-2397-y
- Terrill J.R., Al-Mshhdani B.A., Duong M.N., et al. Oxidative damage to urinary proteins from the GRMD dog and mdx mouse as biomarkers of dystropathology in Duchenne muscular dystrophy. PLoS One. 2020;15(10):e0240317. doi: 10.1371/journal.pone.0240317
- Bell C. The nervous system of the human body: as explained in a series of papers read before the Royal Society of London. Edinburgh: Adam & Charles Black, 1830.
- Semmola G. Sopra due malattie. Notizie dell’atra infermita. Acad Pontaniana. 1834;164–5.
- Conte G., Gioja L. Scrofola del sistema muscolare. Annali Clinici dell’Ospedale degli Incurabili di Napoli. 1836;2:66-79.
- Little W.J. On the Nature and Treatment of the Deformities of the Human Frame: Being a Course of Lectures Delivered at the Royal Orthopaedic Hospital in 1843: With Numerous Notes and Additions to the Present Time, London: Longman, Brown, Green & Longmans. 1853.
- Meryon E. Practical and Pathological Researches on the Various Forms of Paralysis. London: John Churchill; 1864: 131.
- Duchenne G.B.A. Recherches sur la paralysie musculaire pseudohypertrophique ou paralysie myo-sclerosique. Arch.Gen.Med. 1868;11:5-25.
- Nozoe K.T., Akamine R.T., Mazzotti D.R., et al. Phenotypic contrasts of Duchenne Muscular Dystrophy in women: Two case reports. Sleep Sci. 2016;9(3):129-133. doi: 10.1016/j.slsci.2016.07.004
- Nowak K.J., Davies K.E. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO reports. 2004;5:872-876. doi: 10.1038/sj.embor.7400221
- Lisak R.P., Truong D.D., Carroll W., Bhidayasiri R. International Neurology. Wiley; 2011:222.
- Nigro G., Comi L.I., Politano L., et al. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol. 1990; 26: 271-277.
- Roudaut C., Le Roy F., Suel L., et al. Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation. 2013;128(10):1094-104. doi: 10.1161/CIRCULATIONAHA.113.001340
- Finsterer J., Stöllberger C.. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol. 2008;24(10): 786-792. doi: 10.1016/s0828-282x(08)70686-x
- Verhaart I.E.C., Johnson A., Thakrar S., et al. Muscle biopsies in clinical trials for Duchenne muscular dystrophy — Patients’ and caregivers’ perspective. Neuromuscul Disord. 2019;29(8):576-584. doi: 10.1016/j.nmd.2019.06.004
- Sun C., Shen L., Zhang Z., Xie X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes (Basel). 2020;11(8):837. doi: 10.3390/genes11080837
- Duan D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol Ther. 2018; 26(10): 2337-2356. doi: 10.1016/j.ymthe.2018.07.011
- Golodirsen (Vyondys 53) for Duchenne muscular dystrophy. Med Lett Drugs Ther. 2020; 62(1603):119-120.
- Al-Dosari M.S., Gao X. Nonviral gene delivery: principle, limitations, and recent progress. AAPS J. 2009;11(4):671-81. doi: 10.1208/s12248-009-9143-y
- Ortez C., Natera de Benito D., Carrera García L., et al. Advances in the treatment of Duchenne muscular dystrophy. Medicina (B Aires). 2019;3:77-81.
- Muntoni F., Tejura B., Spinty S., et al. A Phase 1b Trial to Assess the Pharmacokinetics of Ezutromid in Pediatric Duchenne Muscular Dystrophy Patien
 СМИ зарегистрировано Федеральной службой по надзору в сфере связи, информационных технологий и массовых коммуникаций (Роскомнадзор).
СМИ зарегистрировано Федеральной службой по надзору в сфере связи, информационных технологий и массовых коммуникаций (Роскомнадзор).
Регистрационный номер и дата принятия решения о регистрации СМИ: